DISSOCIATIVE SUBSTANCES
This group consists of chemical compounds that, being antagonists of NMDA receptors, interfere with the signaling relationships of various parts of the brain. The resulting dissociation of internal and external signals is due to the uncompetitive blocking of the effects of glutamic acid, more precisely glutamate anions, which are the main excitatory neurotransmitter for ionotropic glutamate (NMDA) receptors. As a result of discordant influences, the perception of pain, one’s own body and the surrounding environment is altered. Emerging disorders of psychosensory synthesis are accompanied by oneroid-like immersion, memory impairments, and catalepsy phenomena. The effects associated with dissociation are subjectively different from the effects of classical hallucinogens in a sense of alienation, unreality of what is happening. The experience can be either ecstatic or frightening.
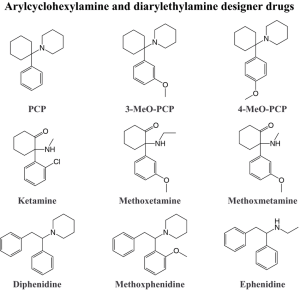
These surfactants of the arylcyclohexylamine group include phencyclidine (English: PCP) and ketamine, which were originally synthesized as anesthetics for surgical interventions, as well as methoxetamine (MXE), a dissociative agent that recently appeared on the illegal market, similar to ketamine and twice its biological activity. In addition, dextromethorphan (DXM), a cough medicine that has dissociative effects in high doses (from 150 mg), is considered in the group of dissociative hallucinogens. The pharmacodynamic properties of dextromethorphan are related to its ability to activate sigma opioid receptors, as well as to the effects of its main metabolite dextrophan (DXO), an NMDA receptor antagonist.
If we do not consider subjects who find pleasure in the sensations of weightlessness, sensory isolation, and total distortion of perception, it remains unclear how substances in this group provide the rewarding effects necessary for addiction to occur. However, animal experiments with intravenous self-administration of phencyclidine, ketamine, and dextromethorphan indicate that similar effects exist.
Phencyclidine (PCP), originally intended for intravenous anesthesia, was soon excluded from the pharmacopoeia due to the fact that anesthetic benefits were complicated by prolonged perceptual disorders with psychotic experiences and behavioral disorders. However, these same hallucinogenic properties have made the drug popular on the illegal drug market.
Phencyclidine is produced by underground laboratories in the form of powder (“angel dust”), tablets and solution, therefore it can be used intranasally, orally and parenterally. The drug is usually mixed with other powders (powdered sugar, ascorbic acid), soluble in neutral liquids. Tobacco, mint, or marijuana can be soaked in phencyclidine solution, although smoking them ensures that only 30% of the initial dose of PCP enters the lungs due to pyrolytic cleavage.
With intravenous administration of the drug at a dose of 0.1 mg / kg, psychotropic effects are detected after 1-2 minutes. Phencyclidine is most often used intranasally, while the effect of an outdoor dose of 2-3 mg occurs after half an hour. The duration of the effects of PCP ranges from 4 to 10 hours or longer, while the method of use has little effect on this indicator. The half—life lasts from 8 to 24 hours, for toxic concentrations – from 2 to 4 days. PCP biotransformation occurs in the liver, the metabolites are excreted in the urine, where they are detected within a few weeks after a single dose of an average dose. The maximum plasma concentration is set after 5-15 minutes during smoking and 2 hours after oral administration (bioavailability 50-90%).
The average street dose of the drug is 3-5 mg, doses of 5-15 mg can cause toxic effects lasting up to 2-3 weeks, and doses exceeding 25 mg can be fatal. The range of toxic plasma concentrations is defined as 10-240 ng/ml. Prolonged use is accompanied by the development of tolerance and withdrawal syndrome with general malaise, mood lability, irritability, hypersensitivity, drowsiness and signs of depression.
The state of intoxication resembles alcohol intoxication with an inability to concentrate, analgesia, dizziness, confusion of thoughts, apathy and drowsiness. Coordination disorders, blurred and inconsistent speech are noted. In general, any symptoms observed with altered forms of intoxication may occur, including hallucinatory disorders and bouts of acute sensory delirium. Orientation disorders occur with wandering and forgetting of events. Unlike alcohol intoxication, detachment and depersonalization phenomena are characteristic. The increase in intoxication is accompanied by catatonic phenomena, muscle rigidity and stupor.
Phencyclidine became notorious in the late 70s of the last century due to its use for “marathons” (repeated use for 2-3 days without eating or sleeping) and subsequent inappropriate behavior in public places.
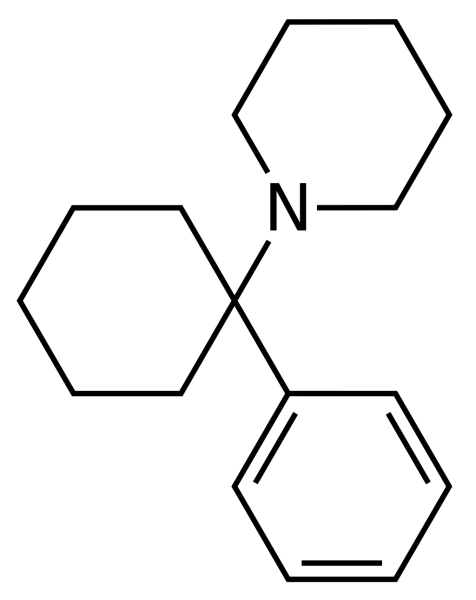
Ketamine was synthesized in the early 60s of the last century as an alternative to phencyclidine. Due to its greater lipophilicity, the drug provides a rapid onset of anesthesia with a shorter duration. Concomitant psychedelic disorders are also characterized by greater transience.
Since the 90s of the last century, ketamine has gained popularity as a “club” drug due to its unusual euphoric effects. The conditions resulting from its use resemble an oneiroid-like syndrome with synesthesia, metamorphosis, a sense of disembodied movement, body schema disorders, and a distorted perception of time and space. In subanesthetic doses, its pseudostimulating effects may be caused by suppression of the inhibitory activity of the cortex. The content of the experiences themselves is largely determined by previous attitudes and the current environment.
Being a non-competitive antagonist of NMDA receptors that blocks the ion channel, ketamine inhibits the neurons of the associative zone of the cerebral cortex and thalamus. As a result, nociceptive afferentation passing through the reticular formation does not reach the cortical regions. Simultaneous stimulation of limbic structures leads to functional disorganization of cortical-thalamic interactions, providing additional effects of dissociative anesthesia. In addition, ketamine in sufficient concentration is able to bind to opioid receptors, which enhances its analgesic activity. Patients look inhibited, while retaining the ability to breathe independently, sometimes they can respond to calls, open their eyes, but do not feel pain impulses, or rather do not perceive them as such. The period of anesthesia and restoration of consciousness is accompanied by amnesia with fragmentary memories of individual psychosensory experiences.
Concomitant anesthesia psychotic disorders, as a rule, are not accompanied by physical activity. In rare cases, however, transient delirious disorders with agitation and aggression are possible. Such conditions are usually preceded by rapidly increasing anxiety and confusion in patients. The psychotic reaction usually lasts no more than
5-7 minutes, exit with full restoration of criticism. At the same time, the memories are fragmentary. Acute ketamine psychosis is associated with excessive release of glutamate and its disturbing activity in certain groups of neurons.
Sympathicotonic effects of ketamine are typical, such as tachycardia, increased blood pressure, and to a lesser extent, lacrimation and salivation. The fact that the hemodynamic effects of the drug do not depend on the dose, and repeated injections may not only not have the same effect, but even cause opposite reactions, makes it difficult to interpret the pharmacodynamic characteristics of ketamine. The phenomena of catalepsy are sometimes accompanied by spasms of the respiratory muscles, trisms and apnea, however, they are quite short-lived and do not require intensive care. Another uncommon side effect of taking ketamine is nausea and vomiting.
In surgical practice, intravenous ketamine administration at a dose of 2 mg/kg leads to anesthesia (mainly somatic) for 30-60 seconds, which lasts 10-15 minutes. It takes about an hour to fully restore orientation. Intramuscular injection is characterized by a slower but longer lasting effect. Intranasal and oral administration of the drug are characterized by even more delayed effects.

Metabolism occurs by demethylation (removal of the metal group — CHZ), while the main metabolic products are excreted in the urine within two to three hours. A small portion of the metabolites may remain for several days, but accumulation does not occur with repeated administration of the drug.
Ketamine has quite pronounced antidepressant properties, however, concomitant hallucinogenic effects and abuse possibilities still limit its regular use in therapeutic practice.
The presence of an official drug makes it possible for parenteral use for non-medical purposes without additional preparations. Ketamine solution can also be ingested (the usual dose is 350-500 mg), but its bioavailability due to active presystemic metabolism does not exceed 20%. At the same time, oral administration of the drug is accompanied by a sufficiently high concentration in the systemic circulation of its main metabolite, norketamine, which has psychoactive properties unrelated to antagonistic effects on NMDA receptors. Thus, the delayed onset and lower intensity of the main effects of ketamine with this method of administration is compensated by a longer psychotropic effect.
Since ketamine is a white crystalline powder in its natural form, it can be consumed intranasally (the usual dosages are 100-400 mg). In the illegal drug market, ketamine is often found in ecstasy pills.
The use of ketamine for addictive purposes is relatively safe in relation to unexpected overdoses, since the drug’s presence on the official pharmaceutical market facilitates its dosing. According to experts from the WHO Committee on Drug Addiction, ketamine abuse does not pose such a significant threat to public health as to justify existing restrictions on its medical use.
However, intravenous ketamine administration leads to rapid onset of derealization and depersonalization effects that persist for one to two hours. In conditions of non-medical use, repeated doses of the drug in combination with other surfactants are often observed. Since ketamine-induced disorders are accompanied by memory impairments, it can be difficult to recall the details of previous anesthesia, as well as the number and name of the additional toxicants used. Prolongation of the toxicological effects of ketamine by other surfactants, in addition to cumulative effects, may be due to metabolic interactions. In particular, since the biotransformation of the drug is carried out by the CYP3A4 enzyme, all its inhibitors act as ketamine synergists.
In general, the picture of ketamine intoxication resembles the abortive course of an LSD overdose with disorientation, kaleidoscopic hallucinations, disorders of consciousness and thinking. In an unusual or threatening environment, experiences can take on the character of extremely unpleasant paranoia. However, sympathomimetic effects are often the reason for hospitalization. Typical, for example, are complaints of chest pain and palpitations, while there is usually no real threat to life.
A study of cases of unintentional overdose, ten times higher than the usual doses used in anesthesiology, demonstrated a fairly long but complete recovery of the condition. There were also no threatening symptoms after repeated repeated use of the drug. Moreover, the neuroprotective properties of ketamine have been used to reduce the damaging effects on the brain cell after traumatic brain injury, stroke, myocardial infarction, epileptic seizures, low oxygen levels and hypoglycemic conditions.
At the same time, there is evidence that regular ketamine users often have urological pathology, in particular, dysuric disorders and cystitis, but opponents consider these data to be an artifact that has no pathogenetic connection with ketamine intake.
In normal clinical practice, the possibilities of laboratory diagnosis of drug abuse are limited, since ketamine is not detected by routine tests.
Toxic damage to neurons as a result of prolonged ketamine use in humans has not yet been confirmed. However, despite the wide enough boundaries of safe use, there may be cases of risky sexual behavior, traffic and household injuries, and other negative events caused by the dissociative effects of ketamine.
Methoxetamine (MXE) acts as an NMDA receptor antagonist while inhibiting dopamine reuptake. It can be used orally and parenterally. The pharmacological effects, as with all arylcyclogexylamines, are determined by the dose. Small doses (10-20 mg) are accompanied by stimulating effects with euphoria and mild coordination disorders, medium doses (20-50 mg) are characterized by agitation, increasing anxiety and psychedelic symptoms, large doses (50-100 mg) lead to total dissociation with perceptual disorders. When ingested, psychotropic effects occur within 10 minutes, reaching a peak within half an hour. On average, there are persistent signs of intoxication for 1.5-2 hours, and it takes about the same amount of time to restore the condition.
The general toxicodynamics of arylcyclohexylamines (PCP, ketamine, and MXE) is characterized by an increase in symptoms from sensory confusion with psychedelic experiences to constipation and coma (sometimes with convulsive seizures). With positive dynamics, the reverse sequence of clinical manifestations is noted. Chronic arylcyclohexyl intoxication can be complicated by dysphoria, deterioration of memory and cognitive abilities, irritability and apathy.
Dextromethorphan (DXM) is the D-isomer of methorphan and has antitussive properties, unlike the L-isomer, which has predominantly opioid analgesic properties. In terms of its ability to suppress the cough reflex, DXM is comparable to codeine, while not having the other effects of opioids. The drug is included in some cough syrups, capsules and tablets. Recommended doses for therapeutic use are 10-20 mg 4 times a day. For recreational purposes, the doses are exceeded a hundred times and usually range from 300 to 1,500 mg. Dextromethorphan abuse is more common among adolescents.
Despite its low toxicity, several deaths associated with dextromethorphan have been documented. However, the role of additional ingredients contained in ready-made dosage forms and used together with dextromethorphan in huge dosages remains unclear.
In therapeutic doses, DXM has no psychotropic effects. In cases of abuse, dextromethorphan itself exhibits pronounced serotonin-ergic properties, and the effect of its main metabolite dextrophan (DXO) is due to the suppression of NMDA receptor activity. Thus, DXM directly causes psychotomimetic effects, while DXO determines its dissociative properties and, ultimately, belonging to the group of hallucinogens.
The conversion of DXM to DXO is carried out using the enzyme CYP2D6. An innate deficiency of this enzyme (in about 5% of the population) or temporary blocking by other drugs can significantly increase the level of DXM relative to DXO.
One of the metabolites of DXM, (H)—methoxymorphinan, also has the ability to block the activity of CYP2D6. As a result, the higher and more frequent the doses of the drug follow one another, the greater the relative amount of DXO relative to DXM. That is, the more pronounced dissociative effects are. Prolonged administration in small doses corresponds to a smaller transformation of DXM into DXO and a change in the balance of psychotomimetic and dissociative effects towards a decrease in the latter. Thus, the addictive effects at different dosages can vary from predominance of euphoria (DXM) to pronounced dissociation (DXO).
At the same time, the use of DXM while taking MAO inhibitors, SSRI antidepressants or other inducers of serotonergic neurotransmission can provoke the development of serotonin syndrome.
Dextromethorphan. It is easily absorbed from the gastrointestinal tract. Symptoms of intoxication occur 15-30 minutes after ingestion of a sufficient dose. The peak concentration is reached on average after
2.5 hours. The genetic polymorphism of the CYP2D6 enzyme activity largely determines the pharmacokinetic characteristics of the drug. If in fast metabolizers the half-life of DXM from plasma takes a little more than 3 hours, in about 10-15% of the subjects this period is prolonged for a day.
In addition to the fact that the nature of psychotropic effects largely depends on the dose of DXM, the subjective perception of the drug’s effects differs significantly from person to person. For about one third, these effects turn out to be attractive, for the other third, on the contrary, unacceptable due to the predominance of dysphoric sensations, and another third leaves this experience completely indifferent. But even adherents of DXM rarely consume it more than once a month. In general, more positive assessments of psychedelic experiences are associated with greater emotional and cognitive involvement of consumers in existential issues.